Синдром кошачьего крика
 Синдром кошачьего крика (также известен под названиями: синдром делеции короткого плеча 5 хромосомы, 5р синдром или синдром Лежена) - редкое генетическое заболевание, связанное с отсутствием части 5 хромосомы. У больных детей с этим заболеванием (преимущественно, но нельзя сказать, что у всех) проявляется плач, который похож на кошачий крик, именно поэтому этот синдром получил название Cri-Du-Chat Syndrome, что происходит от французских слов (плач кошки или крик кота). Впервые болезнь описал Жером Лежен в 1963 году. Частота возникновения синдрома - 1 ребенок на 50000 родившихся, встречается во всех этнических групп и чаще ею болеют женщины, соотношение мужской и женской стати составляет 4:3.
Синдром кошачьего крика (также известен под названиями: синдром делеции короткого плеча 5 хромосомы, 5р синдром или синдром Лежена) - редкое генетическое заболевание, связанное с отсутствием части 5 хромосомы. У больных детей с этим заболеванием (преимущественно, но нельзя сказать, что у всех) проявляется плач, который похож на кошачий крик, именно поэтому этот синдром получил название Cri-Du-Chat Syndrome, что происходит от французских слов (плач кошки или крик кота). Впервые болезнь описал Жером Лежен в 1963 году. Частота возникновения синдрома - 1 ребенок на 50000 родившихся, встречается во всех этнических групп и чаще ею болеют женщины, соотношение мужской и женской стати составляет 4:3.Признаки и симптомы
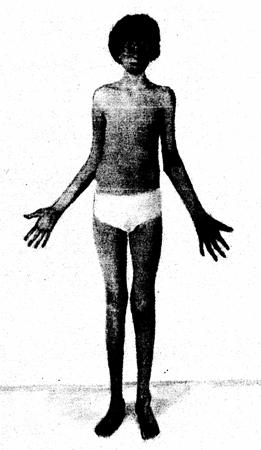
Как уже было отмечено, синдром получил свое название из-за характерного плача детей (он аналогичен мявканню котенка, крика кошки), страдающих этим заболеванием. Это крик происходит из-за проблем с гортанью и нервной системой. Около 1/3 детей теряют эту особую характерную черту до 2 лет. Другими симптомами, которые указывают на заболевания синдромом кошачьего крика являются:
• проблемы с питанием в связи с трудностями при глотании и сосании;
• низкий вес ребенка при рождении и низкие темпы развития (в первую очередь физического);
• существенная задержка развития когнитивных, речевых функций и функций движения;
• проблемы с поведением, такие как: гиперактивность, агрессия, истерики и однообразные движения, постоянно повторяются;
• нетипичные черты лица, которые могут со временем исчезнуть или усилиться;
• чрезмерное, неконтролируемое слюноотделение;
• запоры.
Дополнительными типичными признаками заболевания можно назвать: гипотонию, микроцефалию, задержку физического развития, круглое лицо с полными щеками, гипертелоризм, эпикантус, опущенные углы глазных щелей, косоглазие, плоскую спинку носа, опущенные углы рта, микрогнатию, низко расположеные уши, короткие пальцы, 4 -х пальцевую ладонную складку и порок сердца (например, дефект межжелудочковой перегородки (вентрикулосептальный дефект), дефект межпредсердной перегородки (атриосептальный дефект), открытого артериального протока, тетраду Фалло). У людей с синдромом кошачьего крика обычно нет проблем с половой системой и репродуктивностью (рождением детей).
В позднем детстве и подростковом возрасте выводы врачей включают значительные нарушения умственного развития, микроцефалии, огрубение черт лица, надбровные дуги, глубоко посаженные глаза, гипопластическая перегородку носа, тяжелые нарушения прикуса и сколиоз. Больные девушки достигают половой зрелости, развитие вторичных половых признаков, и менструация у них возникают обычно вовремя. Строение половых органов и путей у лиц женского пола, как правило, нормальное, за исключением двурогой матки, которая местами встречается у этой группы больных. У мужчин яички часто малы, но сперматогенез преимущественно незначительно нарушен.

Синдром кошачьего крика связан с частичной делецией короткого плеча 5 хромосомы, которая также называется "5p моносомия". Примерно 90% случаев этого генетического заболевания являются результатом спорадических или случайных мутаций, то есть новые случаи. Остальные 10-15% возникают из-за неравного разделение родительского генетического материала с нарушением сбалансированной транслокации, где 5р моносомия часто сопровождается соответствующей трисомией этой части генома. Эти лица могут иметь более серьезные проявления заболевания, чем те у кого проявляются изолированные случаи моносомии с 5р (вследствие делеции).
В большинстве случаев заболевание сопровождается полной потерей дистально-расположенной генетической информации, составляющей 10-20% генетического материала на коротком плече пятой хромосомы. Менее 10% случаев имеют другие редкие цитогенетические аберрации (например, интерстициальную делецию, мозаичность, кольца и новые транслокации). Делеция хромосомы 5, родительского происхождения примерно в 80% случаев происходит заново.
Потеря небольшого участка в зоне 5p15.2 (критической для этого заболевания области) коррелирует со всеми клиническими признаками синдрома за исключением кошачьего крика, который возникает при условии нарушений в области 5p15.3 (кошачьей критической области). Полученные результаты свидетельствуют, что две несмежные критические области содержат гены, вовлеченные в этиологию этого заболевания. Два гена в этих регионах, семафорин F (SEMA5A) и дельта Катенин (CTNND2), потенциально участвуют в мозговом развитии. Удаление гена транскриптазы обратной теломеразы (hTERT) локализированной в 5p15.33 может способствовать фенотипическому изменению у больных синдромом кошачьего крика.


 во жизни человека (хотя он не представляет угрозы жизни) является дуральная ектазия. Это ослабление и растяжение твердой оболочки мозга, а точнее соединительной ткани дурального мешка - мембраны, которая окутывает спинной мозг.
во жизни человека (хотя он не представляет угрозы жизни) является дуральная ектазия. Это ослабление и растяжение твердой оболочки мозга, а точнее соединительной ткани дурального мешка - мембраны, которая окутывает спинной мозг. фана болеют как мужчины, так и женщины, при этом нет никаких этнических или географических особенностей данного заболевания. Согласно оценкам ученых один человек из 3000-5000 человек болеет синдромом Марфана. Через аутосомно-доминантный характер заболевания каждый из родителей может передать дефектные гены ребенку с вероятностью 50%. Большинство людей с синдромом Марфана имеют кого то в семье, кто уже поражен этим заболеванием, а 15-30% всех случаев связаны с новыми генетическими мутациями, которые возникают у одного ребенка на 20000 новорожденных. Синдром Марфана - это пример доминирующей негативной мутации и гаплонедостаточности. Вызванной переменной экспрессивностью, тогда как неполная пенетрантность на сегодня не является хорошо задокументированной.
фана болеют как мужчины, так и женщины, при этом нет никаких этнических или географических особенностей данного заболевания. Согласно оценкам ученых один человек из 3000-5000 человек болеет синдромом Марфана. Через аутосомно-доминантный характер заболевания каждый из родителей может передать дефектные гены ребенку с вероятностью 50%. Большинство людей с синдромом Марфана имеют кого то в семье, кто уже поражен этим заболеванием, а 15-30% всех случаев связаны с новыми генетическими мутациями, которые возникают у одного ребенка на 20000 новорожденных. Синдром Марфана - это пример доминирующей негативной мутации и гаплонедостаточности. Вызванной переменной экспрессивностью, тогда как неполная пенетрантность на сегодня не является хорошо задокументированной.